

Cannabidiol (CBD), a primary bioactive phytocannabinoid extracted from hemp, is reported to possess potent anti-tumorigenic activity in multiple cancers.
However, the effects of CBD on bladder cancer (BC) and the underlying molecular mechanisms are rarely reported.
Here, several experiments proved that CBD promoted BC cells (T24, 5637, and UM-UC-3) death.
For example, T24 cells were treated with 12 µM CBD for 48 h, flow cytometry analysis demonstrated that early and late apoptotic cells were accounted for by 49.91%, indicating CBD enhanced cell apoptosis ability.
To deeper explore molecular mechanisms, the CBD-treated T24 cell transcriptome libraries were established.
KEGG analysis implied that the significantly changed genes were enriched in the PI3K/Akt pathway.
qRT-PCR and Western blot assays verified that CBD regulated BC cells growth and migration and induced apoptosis by inactivating the PI3K/Akt pathway.
Meanwhile, the developed chitosan to wrap CBD-loaded PLGA nanoparticles can significantly enhance the adhesion of the material to the mouse bladder wall, and the binding efficiency of mucin to chitosan-PLGA nanoparticles reached 97.04% ± 1.90%.
In summary, this work demonstrates that CBD may become a novel reliable anticancer drug and the developed intravesical adhesion system is expected to turn into a potential means of BC chemotherapy drug delivery.
1. Introduction
Bladder cancer (BC) is the tenth-leading cause of cancer-related deaths around the world [1], with an estimated 55,000 newly diagnosed cases in 2020 globally [2], and approximately 170,000 deaths [3].
At initial diagnosis, almost 75–80% of cases are non-muscle-invasive bladder cancer (NMIBC) and 20–25% patients develop into muscle-invasive disease during their life [4].
At the present stage, intravesical instillation chemotherapy is the mainstay standard of care for high-risk NMIBC after transurethral resection of the bladder tumor [5].
However, there are still two formidable challenges hindering the therapeutic effect.
Chemoresistance gradually occurs in approximately 70% of patients [6]; conversely, interference of bladder physiological characteristics leads to a poor utilization rate of instillation drugs [7].
Therefore, there is an urgent need to find more therapeutic drugs and explore better drug delivery for high-risk NMIBC patients.
In terms of drug discovery, plant-derived products have received considerable attention that possesses characteristics of chemical structural diversity, diverse biologic activities, low adverse reactions incidence, and low costs [8,9].
Notably, it has been extensively studied that diverse anti-cancer compounds from plant sources [10], including vinca alkaloids, taxanes [11], tanshinone [9], and camptothecinsare [12], which are still considered to be the cutting-edge drugs for the treatment of cancers.
For example, paclitaxel, extracted from Taxaceae family, is a well-known natural anti-cancer drug in the past 30 years.
In the clinical treatment of breast cancer, it can cause altered mitosis and cell death by interfering with microtubule function [13,14].
Camptothecin obtained from Camptotheca acuminata (Nyssaceae) has been approved for treatment of colorectal cancer, ovarian cancer, and small cell lung cancer because of its ability to inhibit DNA topoisomerase [15].
Previously, studies above suggested that many plant-derived compounds had good pharmacological properties.
Currently, there is great interest in potential medical use of cannabidiol (CBD) originating from Cannabis sativa (also named hemp), a non-psychoactive phytocannabinoid [16].
Studies have shown that CBD can exert multiple beneficial pharmacological effects, including antiepileptic, anxiolytic, anti-inflammatory, and antipsychotic properties [17].
The CBD drug Epidiolex was approved by the Food and Drug Administration (FDA) in 2018 for the treatment of childhood epilepsy, proving the safety and effectiveness of CBD [18].
A large number of studies have focused on its antitumor activity in different types of tumors.
For example, Soyeon et al. [19] reported that CBD induced apoptosis in colorectal cancer cells by activating Noxa gene expression, and it can promote apoptosis via regulation of XIAP/Smac in gastric cancer cells [20].
Mohamad and his colleagues indicated that CBD possessed antitumor activity mainly by inhibiting the EGF/EGFR pathway in breast cancer [21].
Although CBD has shown good effects in the treatment of a variety of cancers, reports on treatment of bladder cancer are rarely known.
CBD is a potential chemotherapy drug, but how to increase its utilization in the bladder has become a new challenge.
To the best of our knowledge, the residence of free drugs inside the bladder is short due to periodic urination [5].
Given that CBD is highly lipophilic (easy to dissolve in dimethyl sulfoxide (DMSO)) with poor stability (affected by temperature, light, and auto-oxidation [22]), continuous perfusion is required to maintain high drug concentration; thus, this can result in elevated side effects or occurrence of drug resistance.
To solve the existing issues, better drug delivery systems are being developed and applied.
It has been proven that these challenges can be resolved by encapsulating lipophilic drugs in poly-(lactic-co-glycolic acid) (PLGA) particles, thereby allowing extended antitumor activity after a single administration, which is feasible in the long-term treatment of cancer [23].
In addition, chitosan (CS), derived from chitin, has been extensively used as the mucoadhesive materials in the formulation of multiple delivery systems [24].
Furthermore, CS can perform the adhesion function for a long time by reacting with the glycosaminoglycan layer on the surface of the bladder wall [7].
Therefore, coating CS on the surface of PLGA particles is expected to become a novel mucosal delivery system for the treatment of bladder cancer.
In the present study, we respectively investigated the antitumor effects and molecular mechanisms induced by CBD in BC cells and developed a drug delivery system based on positively charged CS coated CBD-loaded PLGA particles.
This work deeply explored the mechanism of CBD-mediated anti-tumorogenesis and laid the foundation for the future development of bladder perfusion drug delivery strategy.
2. Materials and Methods
Reagents and Antibodies. CBD was purchased from Push (Chengdu, China).
The initial concentration of CBD powder dissolved in absolute DMSO was 51 mM and stored at −20 °C in the dark for later use.
When using CBD for follow-up experiments, the DMSO concentration needed to be kept below 1‰. Poly-(lactide-co-glycolic acid-resomer) (PLGA, Mw: 24,000–38,000 Da, i.v. 0.25–0.35 dL/g) was obtained from Daigang (Jinan, China).
Polyvinyl alcohol 1788 (PVA, Mw = 44.05) was purchased from Aladdin (Aladdin, Shanghai, China).
RPMI-1640, DMEM, and DMEM/F12 medium were obtained from Gibco (Thermofisher, Shanghai, China).
Fetal bovine serum (FBS) was supplied by BI (Israel, South America).
Antibiotic-antimycotic solution and 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) were obtained from Solarbio (Shanghai, China).
In situ cell death detection kit and RIPA buffer were purchased from Beyotime (Shanghai, China).
Antibodies against PI3K (1500 μg/mL, 1:10000), Akt (2504 μg/mL, 1:3000), phospho-Akt (1683 μg/mL, 1:6000), mTOR (500 μg/mL, 1:10000), phospho-mTOR (1000 μg/mL, 1:1000), Bcl-2 (1000 μg/mL, 1:2000), Bax (1600 μg/mL, 1:4000), Cytochrome-c (1000 μg/mL, 1:4000), Caspase 7 (700 μg/mL, 1:1000), Erk1/2 (1000 μg/mL, 1:6000), phospho-Erk1/2 (370 μg/mL, 1:3000), MMP9 (400 μg/mL, 1:600), MMP2 (1000 μg/mL, 1:600), and β-actin (427 μg/mL, 1:5000) were purchased from Proteintech (Shanghai, China).
Phospho-PI3K (690 μg/mL, 1:1000) was purchased from Abcolonal (Shanghai, China).
HRP-conjugated secondary antibodies were purchased from Proteintech.
HPLC-grade methanol and acetonitrile, were obtained from Macklin (Shanghai, China).
Cell Culture. Human T24, 5637, UM-UC-3, and SV-HUC-1 cell lines were supported by Sun Yat-Sen memorial hospital (Sun Yat-Sen University), and their biological characteristics are presented in Table S1.
T24 cells and 5637 cells were cultured in RPMI-1640 medium.
UM-UC-3 cells was cultured in DMEM medium, and SV-HUC-1 cells was cultured in DMEM/F12 medium.
All media were supplemented with 10% FBS, and 1% antibiotic-antimycotic solution.
Cells were cultured at 37 °C under a humidified atmosphere of 5% CO2.
Cell Viability Assay. The effect of CBD on the viability of T24, UM-UC-3, 5637, and SV-HUC-1 cells were evaluated quantitatively by the MTT assay.
First, 100 µL of the cell suspension was seeded at a density of 2 × 104 cells per well in 96-well culture plated and incubated overnight.
Subsequently, the medium was changed, each sample well was added with 200 µL volume of different concentrations of CBD (0 and 1‰ DMSO, 10, 15, 20, 25, 30, 40, 50 µM) at 37 °C in CO2 incubator for 48 h.
Then, MTT reagent (20 µL of 5 mg/mL) was added into each well and incubated at 37 °C for an additional 4 h.
Afterward, the supernatant was carefully removed, and the formed formazan crystals were dissolved by adding 150 µL DMSO to each well.
The plates were shaken for 10 min at room temperature; absorbance at 490 nm was measured by using the automatic microplate reader (BioTek, Winooski, Vermont, USA).
Colony Formation Assay. One thousand cells were seeded in 6-well plates in complete medium.
The next day the media were changed to fresh medium containing CBD, and after culturing for 48 h, the fresh medium was replaced, and changed every 3 days.
After 2 weeks, the medium was discarded, and the cells were washed twice with PBS.
The cells were fixed with 4% paraformaldehyde for 15 min, and then the fixing solution was discarded.
The cells were then stained with 1‰ crystal violet for 10 min and rinsed with running water slowly.
The plates were then observed for the formation of colonies.
Wound-healing Assay. Cell migration was assessed in bladder cancer cells using a wound healing assay.
When the density reached 100% confluence, the wound was scratched with a sterile 200 μL pipette tip in the confluent monolayer at the center of culture plates, followed by three washes with PBS. Cells were incubated in medium supplemented with 0.1% (v/v) FBS in the presence or absence of CBD or vehicle.
Images of the scratches were captured at 0, 6, 12 and 24 h to visually assess cell migration distance.
The migratory distance was detected using Image J software (V1.8.0.112, National Institutes of Health, Bethesda, USA).
Hoechst 33258 Staining Assay. The nuclear morphological changes in CBD-treated bladder cancer cells were evaluated using the Hoechst 33258 stain (Solarbio, Shanghai, China). Briefly, equal number of cells was seeded in 6-well plates overnight.
Cells were then washed twice with PBS and stained for 10 min with 10 µg/mL Hoechst 33258 at 37 °C in the dark after treating the cells with CBD or vehicle for 48 h.
Then dyeing medium was removed and the wells were washed twice more with PBS, and the nuclear morphology of the cells was observed under a fluorescence microscope (Olympus, Tokyo, Japan).
Terminal Deoxynucleotidyl Transferase (TdT) dUTP Nick-End Labeling (TUNEL) Assay.
TUNEL assay was used to detect apoptosis and was performed using in situ Cell Death Detection Kit (Beyotime, Shanghai, China).
Bladder cancer cells were seeded in confocal dishes at 37 °C.
After 24 h of culture, culture medium containing 12 µM of CBD was added and further cultured for 48 h in the incubator.
Cells were washed twice with PBS and fixed with 4% paraformaldehyde for 30 min.
Cells were then permeabilized with PBS containing 0.3% Triton X-100 at room temperature for 5 min.
After washing the cells three times with PBS, TUNEL reaction mixture from the assay kit was used to co-incubate with cells for 1 h at 37 °C in a 5% CO2 incubator.
The dishes were washed with PBS and stained with 10 µg/mL Hoechst 33258 for 10 min at 37 °C.
These cells were observed by confocal microscopy (Olympus, Tokyo, Japan).
Apoptosis Assay. The Annexin V-FITC/Propidium Iodide (PI) (Elabscience, Wuhan, China) double staining assay was performed according to the manufacturer’s manual.
Briefly, bladder cancer cells were seeded at a density of 5 × 105/well in 6-well plates overnight and then stimulated by 1‰ DMSO or 12 µM CBD as the vehicle control or experimental group.
After treating for 48 h, the cells and culture medium were harvested using trypsin-EDTA and centrifuged at 500 rpm for 5 min at room temperature.
The supernatant was discarded, and pellet was resuspended in PBS.
Cell suspension was centrifuged under the same condition.
The supernatant was discarded and resuspended by adding 100 µL of 1× binding buffer.
Then, 2.5 µL of Annexin V-FITC and PI staining solution were added to untreated and treated cell suspension, mixed well, and incubated for 15 min at room temperature without light.
Finally, 100 µL of 1× binding buffer was added to the suspension again.
Fluorescence intensity was measured using the Beckman flow cytometer (Beckman Coulter, Inc.250 S.Kraemer Boulevard Brea, CA 92821, USA), and the apoptotic rates of CBD-treated cells were analyzed by using FlowJo software (FlowJoV10, Becton, Dickinson & Company, New York, NY, USA).
RNA Sequencing (RNA-seq) Assays.
The extracting RNA from CBD-treated cells (Figure S3) were used to RNA-seq.
The transcriptome libraries were constructed by Shanghai Majorbio Technology Co., Ltd (Shanghai, China).
All quality inspection data were presented in Table S2. The mRNA expression data was uploaded to the DEseq2 software website (https://login.majorbio.com/login, accessed on 7 September 2021).
To identify differential expression genes (DEGs) between two different samples, the expression level of each transcript was calculated according to the transcripts per million reads (TPM) method.
In addition, Kyoto Encyclopedia of Genes and Genomes (KEGG) functional-enrichment analysis was performed to identify which DEGs were significantly enriched in metabolic pathways at Bonferroni-corrected p value ≤ 0.05 compared with the whole-transcriptome background.
KEGG pathway analysis was conducted by Goatools (https://github.com/tanghaibao/Goatools, accessed on 7 September 2021) and KOBAS (http://kobas.cbi.pku.edu.cn/home.do, accessed on 7 September 2021).
Quantitative Real-time PCR Analysis.
Total RNAs extraction kit, the First-Strand cDNA synthesis kit, and qRT-PCR kit were purchased from Promega (Shanghai, China).
All the indicated samples were normalized to actin and then relative mRNA levels were calculated by using the comparative Ct method.
The qRT-PCR primers (Table S3) were synthesized by Sangon (Shanghai, China).
Western Blotting Analysis.
Cells were lysed by Radio Immunoprecipitation Assay (RIPA) buffer with 1 mM PMSF to obtain the proteins for Western blotting.
The BCA Protein Assay Kit (Sangon, Shanghai, China) was used to quantify the protein concentration.
Subsequently, Western blotting experiments were performed in accordance with the previously reported laboratory procedures [25].
Preparation of CS-coated CBD-loaded PLGA Nanoparticles. Bernhard Brauner et al. [26] had been confirmed that PLGA nanoparticles (PLGA NPs) are more suitable than PLGA microparticles for instillative therapy.
Hence, following a solvent evaporation technique, PLGA NPs loaded with CBD were prepared from an oil-in-water emulsion.
Briefly, PLGA (25 mg) and CBD were dissolved in 2 mL of mixed reagent (Vacetone:Vabsolute ethanol = 4:1).
The oil phase was added dropwise to 20 mL of a 1% (w/v) PVA aqueous solution under stirring at 800 rpm in a magnetic stirrer for 5 h to allow solvent evaporation.
Then, the NPs were collected by centrifuge and washed with 10 mL of ultrapure water three times in order to eliminate remnants of PVA, namely CBD-loaded PLGA NPs (CBD/PLGA NPs).
Next, samples were resuspended in 10 mL ultrapure water and subsequently poured into equal volume of 0.16% (w/v) CS solution and stirred at 800 rpm in a magnetic stirrer for 2 h at room temperature.
Finally, 500 µL ultrapure water was added as a cryoprotectant, and samples were frozen at −80 °C overnight and freeze-dried for 24 h using a freeze dryer, namely CS-coated CBD-loaded PLGA NPs (CS-CBD/PLGA NPs).
For the internalization and adhesion experiments, DiI (Beyotime, Shanghai, China) was used as the fluorescent agent, DiI-loaded PLGA NPs (DiI/PLGA NPs) and CS-coated DiI-loaded PLGA NPs (CS-DiI/PLGA NPs) were prepared using the aforementioned protocol.
Characterization of CS-CBD/PLGA NPs.
The morphologies of NPs (Blank PLGA NPs, CBD/PLGA NPs and CS-CBD/PLGA NPs) were observed using transmission electron microscope (TEM) (JEM-1400, JEOL, Tokyo, Japan).
For TEM examination, the lyophilized NPs were dissolved and placed onto formvar-coated copper grid and negatively stained with phosphotungstic acid.
The size distribution, zeta potential, and PDI of NPs were measured by dynamic light scattering technique (DLS) (Malvern, Malvern city, UK). Each sample was measured in triplicate.
Determination of EE. The lyophilized NPs (CBD/PLGA NPs and CS-CBD/PLGA NPs) were dissolved in 1 mL DMSO for 6 h and then were centrifuged for 30 min at 4 °C at 14,000 rpm.
The supernatant was collected and determined by high performance liquid chromatography (HPLC) (Agilent 1200 series, Agilent Technologies Inc., CA, USA ) equipped with a reverse-phase C18 (250 × 4.6 mm, 5 µm) column.
The mobile phase consisting of acetonitrile (A) and water (B) was used for separation of CBD through isocratic elution at a flow rate of 1 mL/min and an injection volume of 10 µL.
CBD detection and quantification wavelength was set at 220 nm.
The EE was calculated using the following formula.
EE(%)=Detected amount of CBD in the NPs 𝐼𝑛𝑖𝑡𝑖𝑎𝑙 𝑎𝑚𝑜𝑢𝑛𝑡 𝑜𝑓 𝐶𝐵𝐷 𝑖𝑛 𝑡ℎ𝑒 𝑁𝑃𝑠×100%
Determination of Fourier Transform Infrared (FTIR) Spectroscopy.
FTIR spectra of samples homogeneously mixed with KBr and compressed into discs were traced in the range of 400 to 4000 cm−1, using FTIR spectrophotometer (Shimadzu, Kyoto, Japan).
Stability Studies. The different formulations were stored at 4 and 25 °C for a period of 5 weeks.
Samples were taken after 0, 2, and 5 weeks and the following physicochemical properties were examined, including particle size and encapsulation efficiency (EE,%).
In Vitro Assay of Drug Release Studies.
Briefly, 10 mg of NPs were dispersed in 1 mL of phosphate buffered saline (PBS containing 1% (v/v) Tween 80) with different pH (i. e., 5.0 and 6.5) and then incubated at 37 °C at 100 rpm.
At predetermined time intervals, microcentrifuge tubes containing CBD-NPs in PBS were centrifuged at 12,000 rpm for 10 min at 4 °C, and then collected 100 µL clear solution of supernatants to analyze by HPLC.
The release medium was replaced by fresh dissolution medium at each interval; 100 µL samples were then withdrawn and replenished with fresh media.
In Vitro Assay of Mucoadhesion Studies.
The adsorption–association of mucin with the particles was used as a method to assess mucoadhesive properties of the particles prepared [27].
Here, 2 mL of mucin suspension (0.5 mg/mL) and 2 mL of 2 mg/mL NPs dispersion (Blank PLGA NPs and CS-PLGA NPs) was mixed (vortexed) and incubated in a shaker at 37 °C at 100 rpm for 2 h.
The mixtures were then centrifuged at 14,000 rpm for 30 min, and the supernatant was collected and used for the measurement of free mucin.
Bradford reagent is an effective means to detect the concentration of free mucin to further evaluate the amount of mucin adsorbed on the NPs.
According to the Bradford standard curve, 10 µL of all samples (of known and unknown mucin concentration) are mixed with 190 µL Bradford reagent, and then used to detect the absorbance at a wavelength of 595 nm. Finally, the mucin content of the samples was calculated and determined.
In Vivo Assay of Mucoadhesion Studies. Mucoadhesion test was performed according to the previously described method by Martin et al. [28].
Mice were anaesthetized by intraperitoneal injection of phenobarbital sodium.
External genitalia were cleansed and urine in their bladders were emptied.
The bladders were then filled with PBS with or without CS-modified, DiI-loaded NPs (2 mg/mL DiI/PLGA NPs and CS-DiI/PLGA NPs) for 2 h.
Mice were sacrificed, and bladders were washed extensively with PBS to remove non-adherent NPs.
Samples were frozen, sliced, and captured with CLSM.
Cellular Uptake Analysis. T24 cells with density of 5 × 105 cells/well were grown on confocal dishes overnight.
A volume of 100 µL NPs including DiI/PLGA NPs and CS-DiI/PLGA NPs was incubated for 2 h and 6 h at 37 °C.
Then, the excess NPs were washed by fresh PBS for three times.
The cells were fixed by 4% paraformaldehyde for 15 min at room temperature, washed three times with PBS, and then incubated with 10 µM Hoechst 33258 dye for 5 min.
After three washes with PBS, cells were captured by confocal laser scanning microscope (CLSM, Nikon, Shanghai, China).
Statistical Analysis. All data are presented as the mean ± standard deviation (SD) of the mean following analysis with GraphPad Prism 5.0 (GraphPad Software, San Diego, CA, USA).
Statistical significance values were evaluated through one-way ANOVA test with post hoc contrasts by Student–Newman–Keuls test, or part of the data were conducted by Student’s t test, using SPSS 22.0 software for evaluation. p < 0.05 was defined as statistically significant difference.
3. Results and Discussion
3.1. CBD Inhibited Cell Viability and Induced Apoptosis in BC Cells
In order to evaluate the effect of CBD (Figure 1A) on BC in vitro, the MTT assay was performed by using different CBD concentrations for 48 h in various bladder cells, including cancer cells T24, UM-UC-3, 5637, and normal bladder cells (NBC) SV-HUC-1.
As shown in Figure 1B, CBD (0–25 µM) inhibited the viability of bladder cells in a concentration-dependent manner, except for the NBC SV-HUC-1.
The half maximal inhibitory concentration values (IC50) were calculated: 10.85 ± 2.18 µM, 21.83 ± 1.32 µM, 22.92 ± 0.97 µM, 40.68 ± 1.87 µM for T24, UM-UC-3, 5637, and SV-HUC-1 cells, respectively.
As a result, T24 cells were more sensitive to CBD compared with other cell lines.
To examine the effect of CBD treatment on clonogenic survival, the cell colony formation assay was performed.
This ability was reduced by CBD treatment, especially for T24 cells (Figure 1C,D).
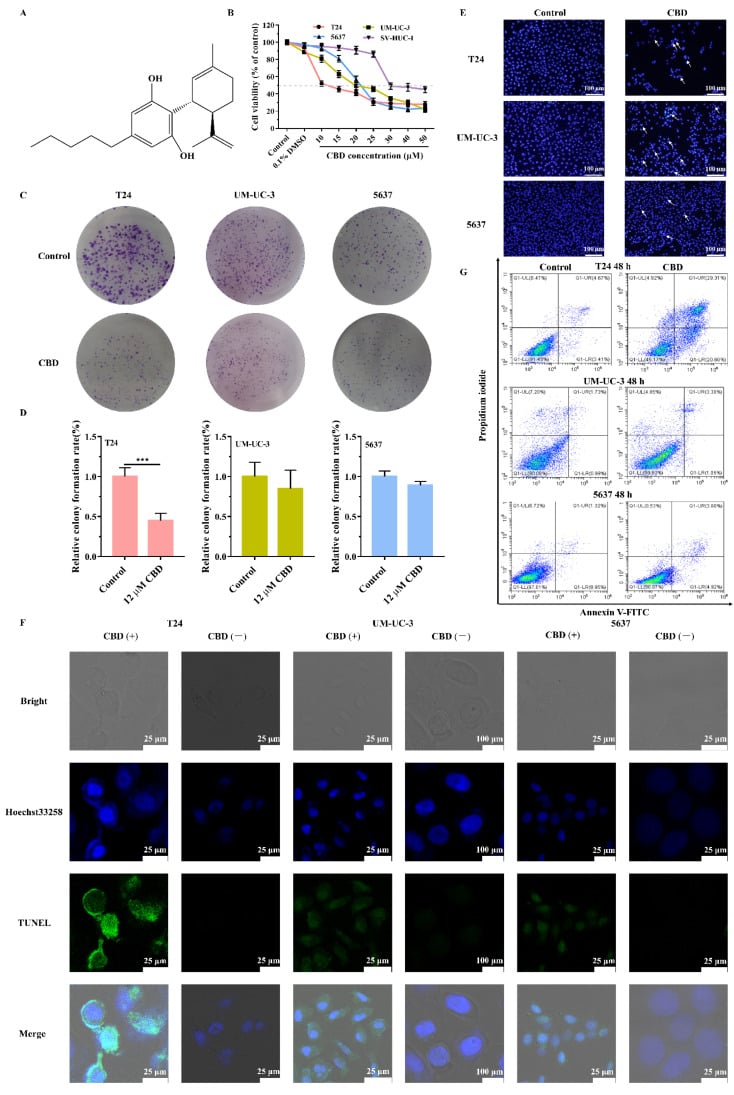
Effect of CBD on BC cells viability and apoptosis.
(A) Chemical structure of CBD.
(B) NBC (SV-HUC-1) and various BC cells were treated with 0 and 1‰ DMSO, 10, 15, 20, 25, 30, 40, 50 µM of the CBD for 48 h.
Cell proliferation was examined by MTT assay.
(C) BC cells were treated with 0 or 12 µM of CBD.
After 2 weeks, the cells were stained with crystal violet, and the colonies were photographed using a digital camera.
(D) Statistical analysis of the colony rate of BC cell population after CBD treatment.
(E) Nuclear fragmentation and condensation (Hoechst 33258 staining) were detected by fluorescence microscopy (magnification, ×100) after treatment with 12 µM CBD for 48 h.
All of these signs are representative of apoptosis.
(F) Cell apoptosis was detected with TUNEL assay, and Hoechst33258 was used as a co-stain to dye the nuclei of the BC cells.
After treatment of CBD (12 μΜ), damaged DNA was visualized in bright green (TUNEL-positive cells), indicating apoptosis.
(G) Cells stained with Annexin V-FITC and PI were studied using flow cytometry to quantitatively detect the apoptosis induced by exposure to 12 μM CBD in BC cells.
Values are shown as mean ± SD of three independent experiments. *** p < 0.001 compared to the control.
